EC145, in combination with pegylated liposomal doxorubicin (Doxil®/Caelyx®) in patients with platinum-resistant ovarian cancer, met its primary endpoint by showing an 85 percent (2.3 month) improvement in median progression-free survival in the intent-to-treat population, and a 260 percent (4.0 month) improvement in a subset of folate receptor positive patients. The final EC145 phase 2 clinical study data were presented today at the 2011 American Society of Clinical Oncology Annual Meeting.

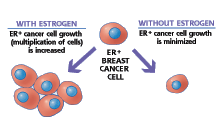
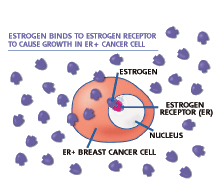
EC145 delivers a very potent vinca chemotherapy directly to cancer cells by targeting the folate receptor expressed on cancer cells, but not on most normal cells. Approximately 80-90 percent of ovarian and lung cancers express the receptor, as do many other types of cancer. Click on the picture above to view a video regarding EC145's mechanism of action. (Photo: Endocyte, Inc.)
Endocyte, Inc., a biopharmaceutical company developing targeted small molecule drug conjugates (SMDCs) and companion imaging diagnostics for personalized therapy, today announced that the phase 2 PRECEDENT trial, which is investigating the company’s lead drug candidate, EC145, in combination with pegylated liposomal doxorubicin (PLD)(brand name: Doxil®/Caelyx®) in patients with platinum-resistant ovarian cancer, met its primary endpoint by showing: (i) an 85 percent (2.3 month) improvement in median progression-free survival (PFS) in the intent-to-treat population, and (ii) a 260 percent (4.0 month) improvement in a subset of folate receptor positive patients. EC145 in combination with PLD showed limited additional toxicity compared to standard therapy with PLD alone. The most commonly occurring adverse events were neutropenia, small intestine obstruction, and palmar-plantar erythrodysesthesia (or hand-foot syndrome). EC145 is a therapeutic that targets the folate receptor and EC20 is a companion imaging diagnostic used to assess folate receptor presence.
These final PFS data from the PRECEDENT trial were presented today at the 2011 Annual Meeting of the American Society of Clinical Oncology (ASCO), in Chicago, Illinois and are available at http://investor.endocyte.com/events.cfm.
“I am encouraged by these data as EC145 is the first drug candidate to demonstrate a significant improvement in PFS in a randomized trial of patients with platinum-resistant ovarian cancer, a very challenging disease with a high unmet need. Historical data show that for these patients on standard therapy, PFS is approximately three months and overall survival is approximately twelve months. No new drug has been approved in the U.S. for this indication in over a decade,” said Wendel Naumann, M.D., Associate Director of Gynecologic Oncology at Blumenthal Cancer Center, Carolinas Medical Center. “In addition the companion imaging diagnostic, EC20, is designed to identify patients who over-express the targeted receptor and are most likely to respond to EC145. This represents a personalized therapeutic approach that I believe will help oncologists direct patients to the most promising treatment.”
EC145 Phase 2 PRECEDENT Clinical Study Data
The Phase 2 PRECEDENT trial was an international, multi-center, randomized study of 149 women with platinum-resistant ovarian cancer. Patients were randomized to receive EC145 plus PLD or PLD alone at a standard dose until disease progression or death. The primary endpoint of the study was progression-free survival. Secondary endpoints included response rate and overall survival.
The EC145 phase 2 PRECEDENT trial results are summarized below.
- 85 Percent (2.3 Month) Improvement in Median Progression-Free Survival for All Patients
Patients receiving EC145 in combination with PLD, regardless of folate receptor expression, had a median progression free survival of 5.0 months compared to 2.7 months for patients receiving single agent PLD. The hazard ratio for PFS was 0.626 (p=0.031). The overall response rate was 28 percent in the EC145 combination group versus 16 percent in the PLD-alone group. CA-125 (cancer antigen-125) responses were also more common in patients receiving EC145 in combination with PLD compared to those receiving PLD alone, with response rates (based on CA-125 blood serum measurement) of 38 percent and 19 percent, respectively.
- 260 Percent (4.0 Month) Improvement in Median Progression-Free Survival for Patients Most Positive for Folate Receptor
The study also utilized EC20, an investigational companion imaging diagnostic that is designed to identify patients with folate receptor positive tumors. As expected, in the folate receptor positive patients the improvement in PFS was even greater. In the subpopulation most positive for the folate receptor, PFS increased 4.0 months from 1.5 months to 5.5 months. The hazard ratio for PFS was 0.381 (p=0.018). This represents more than a 60 percent reduction in the risk of progression and provides evidence supporting the mechanism of action through targeting of the folate receptor. The overall response rate was 22 percent in the EC145 combination group versus 7 percent in the PLD alone group. CA-125 responses were also more common in patients receiving EC145 in combination with PLD compared to those receiving PLD alone, 43 percent and 13 percent, respectively.
“We see tremendous potential for continued development of EC145 based on these data that demonstrate, for the first time, a significant improvement in PFS in patients with platinum resistant ovarian cancer,” said Ron Ellis, President and Chief Executive Officer of Endocyte. “We have already advanced EC145 into Phase 3 evaluation with the PROCEED trial, which has a similar design to the PRECEDENT trial, and plan to file for regulatory approval in the European Union based on the PFS data from the PRECEDENT study.”
Plan to File PRECEDENT Data for Conditional Approval in Europe
As a result of Endocyte’s interaction with the European Medicines Agency (EMA), including a meeting with the Scientific Advice Working Party and written advice from the Committee for Medicinal Products for Human Use (CHMP), the Company will prepare marketing applications for both EC145 and EC20. Based on feedback from the CHMP, Endocyte plans to seek conditional marketing authorization for patients with platinum-resistant ovarian cancer who test positive for the folate receptor using the EC20 companion imaging diagnostic.
Phase 3 PROCEED Trial Initiated
 Endocyte recently announced the initiation of patient enrollment in the Phase 3 PROCEED trial of EC145, which is structured to replicate the PRECEDENT trial design. The Phase 3 trial is a randomized, double-blinded trial of EC145 in combination with PLD compared to PLD plus placebo. The patient population — those with platinum-resistant ovarian cancer — will be the same as in the PRECEDENT trial, and the primary endpoint will be progression-free survival in patients selected by EC20 as folate-receptor positive. The trial will also be statistically powered for overall survival as a secondary endpoint with projected enrollment in excess of 500 patients. The trial will be conducted in approximately 150 sites in the U.S., Canada, and Europe. More information regarding the trial is available at www.clinicaltrials.gov.
Endocyte recently announced the initiation of patient enrollment in the Phase 3 PROCEED trial of EC145, which is structured to replicate the PRECEDENT trial design. The Phase 3 trial is a randomized, double-blinded trial of EC145 in combination with PLD compared to PLD plus placebo. The patient population — those with platinum-resistant ovarian cancer — will be the same as in the PRECEDENT trial, and the primary endpoint will be progression-free survival in patients selected by EC20 as folate-receptor positive. The trial will also be statistically powered for overall survival as a secondary endpoint with projected enrollment in excess of 500 patients. The trial will be conducted in approximately 150 sites in the U.S., Canada, and Europe. More information regarding the trial is available at www.clinicaltrials.gov.
About EC145
EC145 is a conjugate of the vitamin folate and a super-potent vinca alkaloid. Folate is required for cell division and rapidly dividing cancer cells often over-express folate receptors in order to capture enough folate to support cell division. By attaching a chemotherapy drug to folate through proprietary chemistry, EC145 targets cancer cells while avoiding most normal cells. This targeted approach is designed to provide treatment with super-potent drugs while lowering toxicity compared to standard chemotherapy.
About EC20
EC20 is a folate-targeted molecular imaging agent that is being developed as a non-invasive method to identify tumors that over-express folate receptors. These tumors are the molecular target of Endocyte’s folate-targeted therapeutic compounds such as EC145. To date, EC20 has been administered to over 500 patients and has been found to be well tolerated.
About Endocyte
Endocyte is a biopharmaceutical company developing targeted therapies for the treatment of cancer and inflammatory diseases. Endocyte uses its proprietary technology to create novel small molecule drug conjugates (SMDCs) and companion imaging diagnostics for personalized targeted therapies. The company’s SMDCs actively target receptors that are over-expressed on diseased cells, relative to healthy cells. This targeted approach is designed to enable the treatment of patients with highly active drugs at greater doses, delivered more frequently, and over longer periods of time than would be possible with the untargeted drug alone. The companion imaging diagnostics are designed to identify patients whose disease over-expresses the target of the therapy and who are therefore more likely to benefit from treatment.
Sources:
- Naumann RW, Coleman RL, Burger RA, et. al. PRECEDENT: A randomized phase II trial comparing EC145 and pegylated liposomal doxorubicin (PLD) in combination, versus PLD alone, in subjects with platinum-resistant ovarian cancer. J Clin Oncol 29: 2011 (suppl; abstr 5045).
- EC145 PRECEDENT Trial Results — 2011 American Society of Clinical Oncology Annual Meeting Poster Presentation, June 5, 2011. [Adobe Reader PDF document]
- Endocyte’s EC145 Meets Primary Endpoint in Phase 2 Study, Demonstrating 85 Percent (2.3 month) Improvement in Median Progression-Free Survival for Treatment of Platinum Resistant Ovarian Cancer, Press Release, Endocyte, Inc., June 5, 2011. [Adobe Reader PDF document]
- A Randomized Phase II Trial Comparing EC145 and Pegylated Liposomal Doxorubicin (PLD/Doxil/Caelyx) in Combination, Versus PLD Alone, in Subjects With Platinum-Resistant Ovarian Cancer (“PRECEDENT” Trial)(status: enrollment complete), Clinical Trial Summary, ID#: NCT00722592, http://www.ClinicalTrials.gov.
- A Randomized Double-Blind Phase 3 Trial Comparing EC145 and Pegylated Liposomal Doxorubicin (PLD/Doxil®/Caelyx®) in Combination Versus PLD in Participants With Platinum-Resistant Ovarian Cancer (“PROCEED” Trial) (status: recruiting patients), Clinical Trial Summary, ID#: NCT01170650, http://www.ClinicalTrials.gov.
- PROCEED Clinical Trial Website, www.endocyte.com/proceed/
- Mechanism of Endocyte ‘s EC145 Targeted Therapy, Libby’s H*O*P*E* Video Library, http://www.vodpod.com/libbyshopetm, May 26, 2011.

 U.S. SBRT Liver Metastases Study
U.S. SBRT Liver Metastases Study